RESURRECTING DAMAGED NEURONS: ARE WE FIGHTING A HOPELESS BATTLE?
Spinal cord injury (SCI) has been documented throughout history. Early physicians described patients with this affliction to be conscious and aware yet unable to innervate their limbs. Up to the mid 1960s the proper course of treatment for SCI was to stabilize the spinal cord to avoid further injury. Repair, however, was deemed impossible. It wasn’t until recent developments in molecular biology and biotechnology over the past decade that an understanding of the true molecular mechanisms behind SCI has been unraveled to provide a small glimpse of hope. To this day, there is not a cure for SCI but studies in biochemistry and genetics are starting to paint a clearer picture of the events that occur during SCI. With these insights comes the hope that there is a cure buried among the depths of this information. However, there exists some doubt as to whether a cure is really possible. Are scientists and physicians fighting a hopeless battle?
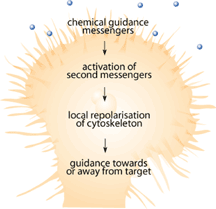
Since the mid 1990s when the first neuronal chemical attractant was identified by Tessier-Lavigne and his group[1-3], numerous studies have identified chemicals that attract and repel growing neurons during development [4-6]. These chemical signals are important because they guide the developing neuron toward its target so that it can innervate specific neurons or muscle fibers. Among the chemicals and receptors being identified, there were some identified as responsible for destroying the path-finding portions of growing neurons known as the growth cone [4-8]. Growth cones are the sensing unit of the growing nerve cell that is designed to sense the surrounding environment in order to help make decisions about which direction to grow [9]. Immediately following SCI, injured nerves have demonstrated the ability to form new growth cones that explore the surrounding environment and try to make connections that have been lost due to the injury [10,11]. Figure 2 shows how a growth cone should function either during development or in the event of nerve damage. Why, then, are previous connections not reestablished?
Severed neurons have difficulty growing and finding their original targets because the environment surrounding these new growth cones is a hostile one [12,13]. The nerve and the surrounding area has sustained significant trauma. As a result, the insulating myelin sheath has usually been torn and there is inflammation of the surrounding area in response to the injury [14,15].
Secondary Damage
Inflammation under normal circumstances is a welcomed primary immune response to injury because the series of events: heat, swelling, redness and pain are all useful to the body [16]. It promotes the recruitment of macrophages and other immune cells [16]. Macrophages are responsible for the clearance of cellular debris, production of toxins to kill any foreign bacteria or viruses as well as the generation of chemicals that promote the healing process [16]. The spinal cord is usually an immune privileged site, which means that immune cells normally do not have access to this area. However, when SCI is sustained, the barrier that separates the nervous system from the rest of the body is damaged and macrophages gain access.
Under these circumstances, inflammation can do more damage than good because the toxins that macrophages create are deadly not only to bacteria and viruses, but to the cells of the nervous system as well. These toxins include reactive oxygen and nitrogen species that are capable of destroying proteins and damaging cellular membranes [12-15]. Also, macrophages will produce and secrete chemicals known as interleukins. Interleukins have the ability to trigger the activation of many intercellular signals by activating nuclear factor kappa B (NFkB)[16-18]. Active NFkB will lead to the transcription and activation of genes that cause cellular decay and programmed cell death, known as apoptosis, to occur. As a result, the normally positive actions of the immune cells turn out to have disastrous effects on the newly injured spinal cord, causing an effect known as “secondary damage” [12-15]. Through the actions of secondary damage, the initial mechanical damage done to the spinal cord and surrounding tissue is made worse by chemical damage due to inflammatory events [14]. Secondary damage will spread outwards from the point of injury killing damaged cells and, eventually, even healthy cells that are adjacent to the injured areas [14].
Growth Cone Collapse
Although the inflammatory response can make things worse, given enough time inflammation does dissipate, the reactive oxygen and nitrogen species are no longer created and the area is allowed to recover to a normal healthy state. Why, then, will the damaged neurons not send out new growth cones across the damaged area to reestablish previously severed connections?
When the nerve is damaged, the surrounding glia cells tend to up-regulate the production of chondroitin sulfate proteoglycans (CSPG)[19]. These proteins are normally responsible for facilitating interactions with cell adhesion molecules and growth factors necessary to maintain a healthy nervous system [19]. However, the over-production of CSPG causes the formation of a blockade that surrounds the broken nerve endings. This is known as the glial scar and has inhibitory effects towards axon regeneration [20]. Embedded in the glial scar are molecules that have been demonstrated in several lines of experiments to have repulsive effects on the growth cone [19,20].
Nogo protein was the first to be identified as having inhibitory effects towards newly developing growth cones [21]. It is a small transmembrane protein that is found on the inner-most layer of the myelin sheath [22]. This protein was identified by isolating protein fractions from rat brain myelin that displayed inhibitory effects on growing growth cones [22]. Through subsequent characterization, an extracellularly exposed region of 66 amino acids known as Nogo-66 was found to be responsible for the inhibitory effects of Nogo [23-25]. Myelin-associated glycoprotein (MAG), a protein initially implicated in the formation and maintenance of myelin sheaths was also identified to have inhibitory effects on neurite (cultured neuron) outgrowth [26,27]. MAG turns out to have dual roles of developmental significance: it can promote the outgrowth of immature neurons and inhibit the growth of mature neurons [28]. Finally, oligodendrocyte myelin glycoprotein (OMgp), a protein involved with the onset of myelination during the development of growing neurons was also identified to have neurite outgrowth inhibitory effects [29,30]. Nogo, MAG and OMgp together are responsible for the majority of the inhibitory effects observed at the site of injury [31].
Contrary to the conventional single ligand to single receptor model, all three of these proteins have strong affinity towards the same receptor known as the Nogo-66 receptor (NgR) [24,30,32]. The binding of just one of these inhibitory proteins to NgR on the newly developed growth cone is enough to cause the initiation of downstream events. Activated NgR has the ability to signal a downstream cascade. This cascade starts with the activation of Rho, one of the 3 main proteins responsible for actin remodeling within the cell [33-35]. Rho has the ability to down-regulate the effects of Rac and Cdc42, causing destabilization of actin filaments and leading ultimately to growth cone collapse [33,34,36]. Once the growth cone has collapsed, the growing nerve axon can no longer grow in that direction until a new growth cone is formed.
Fighting A Hopeless Battle?
Certainly, with the evidence presented it would seem that regenerating a severed spinal cord is an impossible dream. There are layers upon layers of complications that make it difficult for neurons to grow even if they wanted to. But why are there so many obstacles in place? If the regeneration of peripheral nerves from a cut at your finger tip is possible, why is it so difficult to regenerate damage sustained by the central nervous system? Although the answers still elude scientists, progress in molecular medicine has provided important insights. For example, pyrrolidine dithiocarbamate, an anti-inflammatory drug, when administered to rat models of SCI demonstrate that an attenuated inflammatory response following injury will limit the degree of secondary damage sustained [37]. In mice, stopping NgR from binding to its partners demonstrated partial functional recovery following experimental SCI [38]. Ongoing experiments are underway to identify the genetic determinants that are responsible for inhibiting the growth of recovering neurons. Researchers are looking into treatments such as gene therapy, synthetic nerve fibers, surgical grafting approaches and even the use of specialized electromagnetic fields to stimulate the regeneration of damaged nerves [39-42]. With more work and an even greater understanding of the mechanisms mentioned in this article, potential cures are just around the corner. The once impossible battle against spinal cord injury may not be so hopeless after all.
References
1. Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell. 1994 78(3):409-24.
2. Kennedy TE, Serafini T, de la Torre JR, Tessier-Lavigne M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 1994 78(3):425-35.
3. Colamarino SA, Tessier-Lavigne M. The role of the floor plate in axon guidance. Annu Rev Neurosci. 1995 18:497-529.
4. Garbe D, Bashaw G. Axon guidance at the midline: from mutants to mechanisms. Crit Rev Biochem Mol Biol. 2004 39(5-6):319-41.
5. Salie R, Niederkofler V, Arber S. Patterning molecules; multitasking in the nervous system. Neuron. 2005 45(2):189-92.
6. Hinck L. The versatile roles of “axon guidance” cues in tissue morphogenesis. Dev Cell. 2004 7(6):783-93. Review.
7. Barton WA, Himanen JP, Antipenko A, Nikolov DB. Structures of axon guidance molecules and their neuronal receptors. Adv Protein Chem. 2004 68:65-106.
8. Chotard C, Salecker I. Neurons and glia: team players in axon guidance. Trends Neurosci. 2004 (11):655-61.
9. Hippenmeyer S, Kramer I, Arber S. Control of neuronal phenotype: what targets tell the cell bodies. Trends Neurosci. 2004 27(8):482-8.
10. Martin KC. Local protein synthesis during axon guidance and synaptic plasticity. Curr Opin Neurobiol. 2004 14(3):305-10.
11. Ramesh V. Merlin and the ERM proteins in Schwann cells, neurons and growth cones. Nat Rev Neurosci. 2004 5(6):462-70.
12. Beattie MS, Farooqui AA, Bresnahan JC. Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma. 2000 17(10):915-25.
13. Beattie MS, Li Q, Bresnahan JC. Cell death and plasticity after experimental spinal cord injury. Prog Brain Res. 2000 128:9-21.
14. Beattie MS. Inflammation and apoptosis: linked therapeutic targets in spinal cord injury. Trends Mol Med. 2004 10(12):580-3.
15. Kulkarni AP, Kellaway LA, Lahiri DK, Kotwal GJ. Neuroprotection from complement-mediated inflammatory damage. Ann N Y Acad Sci. 2004 1035:147-64.
16. Janeway, CA., Travers, P, Walport, M, Shlomchik, M. Immunobiology. 5th ed. New York and London: Garland Publishing; c2001.
17. Murakami Y, Shoji M, Hirata A, Tanaka S, Yokoe I, Fujisawa S. Dehydrodiisoeugenol, an isoeugenol dimer, inhibits lipopolysaccharide-stimulated nuclear factor kappa B activation and cyclooxygenase-2 expression in macrophages. Arch Biochem Biophys. 2005 15;434(2):326-32.
18. Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl LM, Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol. 2005 15;174(2):1081-90.
19. Matsui F, Oohira A. Proteoglycans and injury of the central nervous system. Congenit Anom (Kyoto). 2004 44(4):181-8.
20. Jain A, Brady-Kalnay SM, Bellamkonda RV. Modulation of Rho GTPase activity alleviates chondroitin sulfate proteoglycan-dependent inhibition of neurite extension. J Neurosci Res. 2004 77(2):299-307.
21. Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron 1988 1:85-96
22. Caroni P, Schwab ME. Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J. Cell Biol. 1988 106:1281-88
23. Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 2000 403(6768):434-9.
24. Fournier AE, GrandPré T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 2001 409:341-46.
25. Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, Michalovich D, Simmons DL, Walsh FS. Inhibitor of neurite outgrowth in humans. Nature 2000 403(6768):383-4.
26. Lai C, Watson JB, Bloom FE, Sutcliffe JG, Milner RJ. Neural protein 1B236/myelin-associated glycoprotein (MAG) defines a subgroup of the immunoglobulin superfamily. Immunol. Rev. 1987 100:129-51.
27. Salzer JL, Holmes WP, Colman DR. The amino acid sequences of the myelin-associated glycoproteins: homology to the immunoglobulin gene superfamily. J. Cell Biol. 1987 104:957-65.
28. DeBallard ME, Tang S, Mukhopadhyay S, Shen Y, Filbin MT. Myelin-associated glycoprotein inhibits axon regeneration from a variety of neurons via interactions with a sialoglycoprotein. Mol. Cell. Neurosci. 1996 7:89-101.
29. Barton WA, Liu BP, Tzvetkova D, Jeffrey PD, Fournier AE, Sah D, Cate R, Strittmatter SM, Nikolov DB. Structure and axon outgrowth inhibitor binding of the Nogo-66 receptor and related proteins. EMBO J. 2003 22(13):3291-302.
30. Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 2002 417(6892):941-4.
31. He Z, Koprivica V. The Nogo signaling pathway for regeneration block. Annu. Rev. Neurosci. 2004 27:341-68.
32. Liu BP, Fournier A, GrandPré T, Strittmatter SM. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 2002 297:1190-93.
33. Dickson BJ. Rho GTPases in growth cone guidance. Curr. Opin. Neurobiol. 2001 11:103-10.
34. Ettienne-Manneville S, Hal A. Rho GTPases in cell biology. Nature 2002 420:629-35.
35. Luo L. Rho GTPases in neuronal morphogenesis. Nature Rev. Neurosci. 2000 1:173-80.
36. Hall A. Rho GTPases and the actin cytoskeleton. Science 1998 279:509-14.
37. La Rosa G, Cardali S, Genovese T, Conti A, Di Paola R, La Torre D, Cacciola F, Cuzzocrea S. Inhibition of the nuclear factor-kappaB activation with pyrrolidine dithiocarbamate attenuating inflammation and oxidative stress after experimental spinal cord trauma in rats. J Neurosurg Spine. 2004 1(3):311-21.
38. Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004 44(3):439-51.
39. Sapieha PS, Peltier M, Rendahl KG, Manning WC, Di Polo A. Fibroblast growth factor-2 gene delivery stimulates axon growth by adult retinal ganglion cells after acute optic nerve injury. Mol Cell Neurosci. 2003 24(3):656-72.
40. Tsai EC, Dalton PD, Shoichet MS, Tator CH. Synthetic hydrogel guidance channels facilitate regeneration of adult rat brainstem motor axons after complete spinal cord transection. J Neurotrauma. 2004 21(6):789-804.
41. Itoh S, Matsuda A, Kobayashi H, Ichinose S, Shinomiya K, Tanaka J. Effects of a laminin peptide (YIGSR) immobilized on crab-tendon chitosan tubes on nerve regeneration. J Biomed Mater Res B Appl Biomater. 2005 7; [Epub ahead of print]
42. De Pedro JA, Perez-Caballer AJ, Dominguez J, Collia F, Blanco J, Salvado M. Pulsed electromagnetic fields induce peripheral nerve regeneration and endplate enzymatic changes. Bioelectromagnetics. 2005 26(1):20-7.
(Images by Jane Philpot)