MUTATIONS IN THE SEED REGION OF WIZARD mIR-96 ARE RESPONSIBLE FOR HEREDITARY PARSELTONGUE HYPERACUSIS

Annals of Praetachoral Mechanics (2016). Vol 2. Advanced online publication. download pdf
ABSTRACT
Parseltongue is a rare hereditary trait that allows wizards to communicate in the language of serpents. Wizards who are parseltongue not only have the ability to speak to serpents (parselmouth), but also possess the ability to hear ultrasonic frequencies produced by the snake (parseltongue hyperacusis). Genome wide association studies (GWAS) have identified scores of genetic variants that contribute to different wizarding phenomenon. MicroRNAs negatively regulate mRNA expression by binding complementary sites in target mRNAs, with the specificity of interaction being crucially dependent on the miRNA seed region. Single nucleotide polymorphisms (SNPs) within the seed region of miRNAs alter target transcript binding energies and as a result, may disrupt or create miRNA/mRNA target pairs. Here we performed a genome-wide screen for SNPs in the seed sequence of wizard miRNAs to determine variants that are either associated with or causal to parseltongue hyperacusis. As a result, we identified 47 miRNAs with SNPs located in their seed region and hundreds of targets that were differentially regulated by the wild type (WT) and SNP allele. We show that a single nucleotide substitution in the seed region of miR-96 induces a significant discordance in promyleocytic leukemia zinc finger (PLZF) transcript targeting. To our knowledge, the SNP (rs19738442) in miR-96 is the first confirmed example of a novel target binding site being created by a SNP within a miRNA seed sequence. We propose that the identified SNP alters the regulatory role of miR-96 in maintaining gene expression profiles in outer hair cells of the inner ear and contribute to parseltongue hyperacusis.
Keywords: micro-RNAs; miRNAs; epigenetics; GWAS; parseltongue hyperacusis; methylation.
Introduction
Parseltongue, known as the language of serpents, is spoken not only by serpentine creatures, but also by a very small minority of wizards. Parseltongue entails both the capacity to speak (parselmouth) as well as hear (parseltongue hyperacusis) in ultrasonic serpent frequencies. The ability to speak parseltongue usually requires the presence of a snake or a serpent emblem; however, some wizards are known to be proficient in the language without the use of serpentine aids [1]. Without being a parseltongue, wizards and muggles are only able to distinguish the lower frequency hissing sounds of snakes and are therefore incapable of learning the most basic word forms of the serpent language. Wizards endowed with the parseltongue phenotype, by contrast, are able to hear frequencies in the 40-50 kHz range where most parseltongue words become discernible [2].
MicroRNA (miRNAs) are short (20-24 nucleotides) non-coding RNAs that play an important gene regulatory role in plants, animals and wizards [3-6]. Most miRNAs are transcribed from their own promoter and originate from distinct independent transcriptional units; however, some miRNAs are transcribed together with a host gene while other miRNA genes are clustered in polycistronic units that share the same promoter [7]. Once processed, miRNAs are loaded onto RNA induced silencing complex (RISC) proteins where they mediate the post-transcriptional repression or cleavage of messenger RNA (mRNA) transcripts [3,4,7,8]. The binding efficacy between the target site of the mRNA (usually found in the 3’ UTR region) and the miRNA itself is determined by a ~7mer seed region located at the 5’-end of the miRNA. Although mutations affecting miRNA seed region do not usually modulate miRNAs biogenesis, they often cause mis-targeting of the miRNA by altering its complementary base pairing requirements [4,6].
Genome wide association studies (GWAS) are powerful techniques employed to survey the genome for nucleotide variants or markers that are associated with certain traits. Recently, with the advent of next generation sequencing technologies, GWAS studies are now able to sequence and discover millions of single nucleotide polymorphisms (SNPs) across whole genomes with massively parallel processing. These discovery driven, hypothesis generating studies are used to uncover SNPs (or markers) that are either causal to or associated with a given wizard phenotype. It has been well established that miRNAs have an expansive and complex regulatory networks in muggles and wizards alike— a single miRNA can target hundreds of transcripts and one transcript may be targeted by several miRNAs [8]. Since miRNAs are key regulators of protein production, it follows that miRNA-related SNPs (including regulatory SNPs affecting miRNA targeting and/or biogenesis) may lead to phenotypic susceptibility through modulation of miRNA activity. To date, many studies have demonstrated that SNPs in miRNAs or their 3’ UTR targets are associated with wizarding prowess5. Even in the muggle world, a number of studies have been performed to systematically link miRNA human polymorphisms to different disease states [9-12].
In this study, we conducted a GWAS to identify miRNA SNPs that are associated with the parseltongue phenotype and that putatively affect miRNA targeting. Here, we propose that a SNP (rs19738442) in the seed region of miR-96 alters the regulatory role of miR-96 in outer hair cells of the inner ear and contributes to parseltongue hyperacuity.
Materials and Methods
Participants
Wizard subjects, aged between 10 and 95 years, were enrolled through the Ministerial Wizarding Register Department from six municipalities in England (Fen, Little Hangelton, London, Godrics Hollow, Wiltshire and Little Whinging) in three stages. In the first stage, geographical criteria were applied; only subjects from an area with a high probability of finding Parseltongue individuals were selected. In the second stage, putative study subjects were invited based on an expert evaluation of parseltongue communities. Finally, the parseltongue identity of the subject in question was confirmed through a discussion involving the subject and the researcher. Briefly, parseltongue selection criteria were as follows: all volunteers completed an extended questionnaire detailing medical history and parseltongue abilities under veritaserum truth serum. Subjects suffering from one or more spells that could potentially affect hearing ability were excluded. All subjects who passed this initial screening underwent an otoscopic investigation before undergoing: i) a pure-tone audiometry test (encompassing the parselmouth frequency range) and ii) a snake charming performance test. Parseltongue exclusion criteria were a minimal audibility curve greater than 15dB and/or failure of the snake charming performance test.
SNP genotyping analysis
Parseltongue patient (n=180) and non-parseltongue patient (n=3200) whole blood samples were collected at St Mungo’s Hospital for Magical Maladies and Injuries under informed, written patient consent and with approval from the Ministry of Magic- department of Magical Education Research Board. Genomic DNA was extracted using standard protocols. The quality of the genomic DNA was assessed by a nano-drop spectrophotometric analyzer and gel electrophoresis. Each sample was genotyped with the Affymetrix GeneChip Human Mapping 100 K array pair (Santa Clara, CA, USA), as described in the GeneChip Human Mapping 100 K Assay Manual.
Quality control in GWAS
In order to avoid systematic biases, we conducted quality control (QC) on the raw genotyping data to filter unqualified samples and SNPs. The first step in QC is transforming raw genotype data into SNP calls. We employed the algorithm CRLMM, which was designed for the affymetrix array and incorporates HapMap information into its framework, to make a call for a SNP of each sample assuming diploids [25]. After calling for all 338 subjects, 458 971 SNPs were used for subsequent processing. SNPs were excluded if they met any of the following conditions:
i. They had a SNP call rate of <95% (in order to ensure that the identification of SNPs are due to genuine sequence level variation rather than errors produced by the underlying sequencing technology). The overall call rate of a sample is equal to the number of SNPs receiving an AA, AB, or BB genotype call divided by the total number of SNPs on the chip.
ii. SNPs had a minor allele frequency of <5%
iii. SNPs did not map to autosomal chromosomes.
SNPs were mapped a reference list of miRNAs downloaded from an online miRNA database (miRBase: http://www.mirbase.org). Association analysis was then performed on these 145 seed region miRNAs using SNPTESTv2 with parseltongue or normal wizard variables as quantitative traits in order to determine statistically significant differences in allele frequencies between the two groups [14].
In silico miRNA target prediction.
Sequences of 3’ UTRs of wizard genes were obtained from the Wizard UCSC genome browser (http://wizardgenome.ucsc.edu). Two prediction tools, Wizo-TargetScan (http://www.WizoTargetScan.org) and miRanda Wizard (http://www.wizmicrorna.org), were used for the identification of candidate miRNA targets.
In vitro biological validation of miR-96 targeting.
To directly identify miRNA targets, miRTRAP was performed as previously described [26]. Briefly, Mouse Embryonic Fibroblasts (MEFs) were transfected with Pso-modified miRNA negative control, miRNA-96, or miRNA-29a according to the manufacturer’s protocol (Invitrogen). After 72 h, cells were exposed to UVA for 10 min (for cross-linking) and total RNA was pulled down using streptavidin beads. RNase-free water was added to release RNA from the beads and RNA was extracted according to the manufacturer′s protocol (Invitrogen). Final RNA samples were used for reverse transcription reactions and qPCR was performed to for transcript quantification.
Luciferase reporters were constructed for all predicted miR-96 target 3’ UTRs by amplifying genomic DNA and cloning it into a vector. siRNAs were designed to mimic wt miR-96 (siR96) and miR-96 containing the parseltongue (siR96+13G>A) mutation, and miR-140 (siR140) as a negative control. Luciferase activity was normalized to protein content measured using the BCA protein assay kit (Pierce) and intensity values was taken using the Omega (BMG Labtech, Ortenburg, Germany) plate reader.
Animals
Targeted transgenesis and knock-in were performed to target an inducible expression cassette specifically to the prestin repressor gene or the miR-96 gene. Targeted knock-in transgenesis was carried out using a Cre-mediated knockin system on mouse embryonic stem cells as previously described [27]. Briefly, we isolated a 9-kb fragment from the mouse Prestin gene, which contains the putative thyroid hormone responsive element [28]. A cassette containing a 1.7-kb tTA coding region with nuclear localization signal (NLS) was inserted 1 kb downstream in intron 3. The final construct was verified by DNA sequencing. An 11.3-kb fragment was injected into the pronuclei of fertilized one-cell eggs from FVB/NJ mice (Due to prominent pronuclei in their fertilized eggs and large litter size, FVB/NJ are commonly used for transgenic injection). Genotyping of the transgenic founder and offspring was determined by PCR. The tTA-positive transgenic mice were mated with mice containing the miR-96 transgene or the PLZF transgene under control of the tet-o promoter. To knock-in each inducible transgene, a co-transfection was performed with selected plasmids and the Cre expression cassette using culture conditions as previously described [29].
Parselmouth Hyperacuity Assays and Audiogram analysis in mice.
To suppress miR-96 and PLZF expression, mice were administered doxycycline (100 μg/ml) in their drinking water. For blotting experiments, inner ears of mice were isolated and outer hair cells were micro-dissected under the direction of a pathologist.
Hearing Threshold Measurement Using Auditory Brainstem Responses (ABR).
Five animals for each mouse strain were used for ABR recordings. Each mouse was anesthetized with a mixture of ketamine and xylazine. ABR thresholds were measured at the ages of 3, 9 and 12 months for all mice and five animals from each strain were used. ABRs were recorded in response to tone bursts of 2, 2.8, 4, 5.6, 8, 11, 16, 22, 32, 40, 50, and 60 kHz using standard procedures previously described [30]. ABR signals were collected with needle electrodes placed subcutaneously at the mastoid prominence.
TWGA data analysis
Information regarding DNA methylation, RNA expression and clinical annotions was obtained from 320 wild-type and 118 slytherin small samples available through The Wizard Genome Atlas Project [24] (TWGA). Gene dosage alterations were assessed using the Affymetrix SNP6.0 platform at the Broad TWGA Genome Characterization Center. Gene-level transcription estimates, in the form of RPKM counts, were obtained using the Illumina HiSeq 2000 RNA sequencing platform by TWGA Genome Characterization Center. Methylation analyses using the Illumina Infinium HumanMethylation450 platform were performed at Johns Hopkins University, University of Southern California, and TWGA genome characterization center. Probes mapping to the miR-96 gene cluster were extracted. Only probes mapping to the promoter region, which are most likely to have an effect on gene expression, were selected for further analysis. The ratio of the intensity of the methylated bead type to the combined locus intensity (termed as beta values (βV)) was calculated to assess the degree of methylation. The difference in probe methylation between Slytherin and non-Slytherin tissues, a delta beta value (dβV), was calculated for each probe. All data dimensions were aligned to clinical metadata also obtained from the TWGA [24]. Correlations between groups were carried out using GraphPad software v6 and matlab. Group comparisons between Slytherin and non-Slytherin samples were done using an analysis of variance (ANOVA). Correlation between gene expression and promoter hypermethylation was assessed through Spearman correlation analysis using matlab software. In all comparisons, a value < 0.05 was considered significant.
Results
Summary of miRNA SNPs indentified from GWAS
After calling for all 3385 subjects (n=180 parseltongue; n=3205 non-parseltongue), our GWAS identified 458 971 SNPs across the genome putatively associated with the parseltongue phenotype. Among the 321 455 SNPs that passed quality control (QC; Materials and Methods), a total of 649 SNPs (including insertion/deletion polymorphisms) mapped to 411 miRNAs. SNPs in pre-miRNAs were then further classified into miRNA seed regions (n=145) or seed flanking regions (n=903) according to their location within the miRNA identified from the HapMap Consortium.
Because allelic variations in miRNA seed regions have been shown to influence transcript targeting [13], we examined only the proportion of SNPs present in miRNA seed sites. Association analysis was then performed on each inputted miRNA seed sequence SNP (n=145) using SNPTESTv2, with parseltongue or normal wizard variables as quantitative traits, in order to determine statistically significant differences in allele frequencies between the two groups [14]. Our search identified 47 unique seed-region miRNA SNPs that are either associated with or causal to parseltongue-related phenotypes. This collection of SNPs was considered for further analysis.
Two miRNA seed SNPs in parseltongue positive patients are predicted to affect auditory acuity in wizards.
To systematically assess the potential impact of SNP allele-status on miRNA/mRNA binding, we combined results of two bioinformatic prediction tools, miRanda Wizard (http://www.wizmicrorna.org) and Wizo-TargetScan (http://www.WizoTargetScan.org). For the 47 SNPs in miRNA seed regions, a total of 2876 mRNA targets were predicted to interact with at least one miRNA in both databases. Next, only targets which were differentially regulated by miRNAs (ie. transcripts exclusively targeted by the parseltongue allele or the wild-type allele) were considered further. A total of 1658 transcripts were tossed because they were targeted by both the parelstongue SNP-miRNA and its respective wild-type allele (Figure 1; blue). The remaining 1218 mRNA transcripts were exclusively targeted by either the parseltongue allele (85, Figure 1; orange) or the wild-type allele (1133, Figure 1; green), but not by both. Regarding differentially targeted transcripts, nearly 93% of transcript targeting was lost upon mutation to the parseltongue predisposing SNP. Interestingly, we found that 7% of the mRNA targets were acquired only by the parseltongue-associated miRNA but were not predicted to interact with the WT miRNA. For example, the number of putative targets of miR-1276 was increased after variation to the parseltongue allele (Figure 1B).
Using a previously described set of genes dysregulated in OHC electromotility and hearing defects [15] (Materials and Methods), we then selected all putative miRNA/mRNA interactions that might influence wizard auditory acuity. Using this list of genes implicated in wizard auditory sensory acuity, our list of 1218 differentially targeted transcripts was filtered down to a total of 13 transcripts targeted by two miRNAs (miR-96 and miR-124). Seven transcripts were regulated by miR-124 and six by miR-96. In all cases except one, the parseltongue miRNA SNP was predicted to disrupt transcript targeting. Interestingly, one target, PLZF (the repressor of prestin) was predicted to be uniquely acquired by the parseltongue miR-96 variant while remaining untargeted by WT allele.
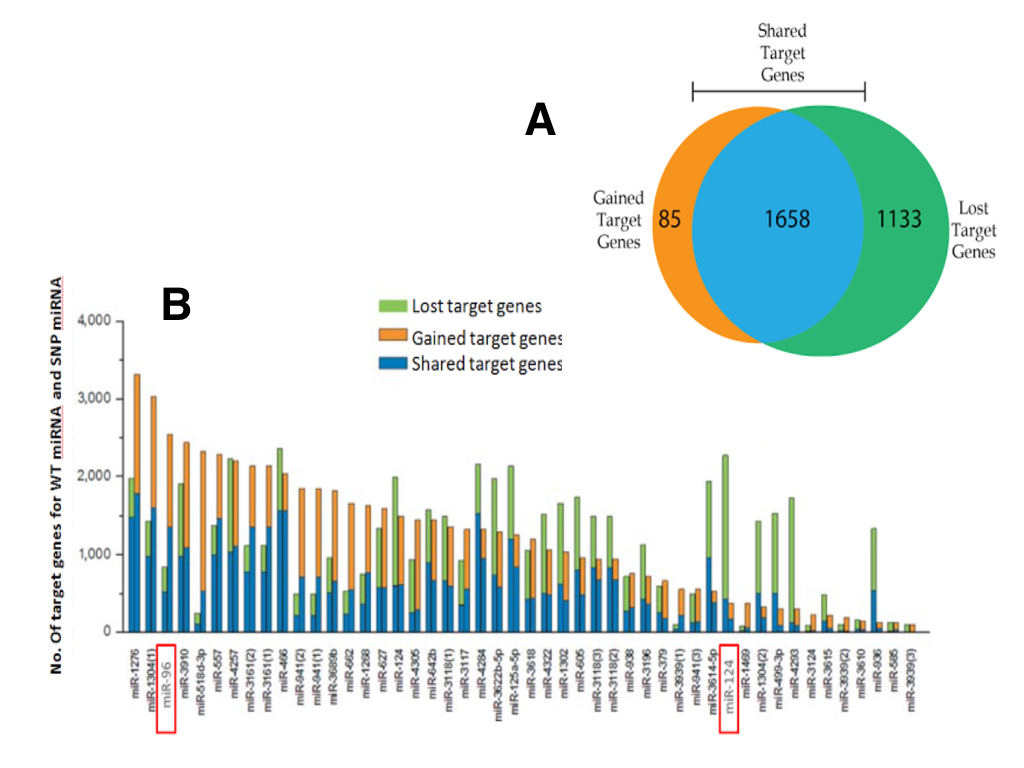
(CLICK IMAGE TO ENLARGE) Figure 1. Target loss and gain analysis of miRNAs by SNP’s in their seed regions. (A) Two tools, miRanda Wizard and Wizo-target scan, were used to predict target genes for wild-type miRNA and SNP-miRNA. These resulted in 3 groups; mRNAs that were not differentially regulated between wild-type miRNA and SNP-miRNA (blue), miRNAs that were preferentially targeted by the wild-type miRNA (green), and miRNAs that were preferentially by the SNP-miRNA (orange). (B) The number of target genes for wild-type miRNAs and for SNP-miRNAs. For each pair of columns for each miRNA, the left column represents the predicted targets for the wild-type miRNA while the right column represents the predicted targets for SNP-miRNA. miR-96 and miR-124 are highlighted as they are the only miRNAs to target mRNA transcripts implicated in wizard auditory sensory acuity.
In vitro validation of miRNA-target pairs shows that the parseltongue predisposing miR-96 variant targets PLZF.
Although SNPs in miR-124 and miR-96 were predicted to alter miRNA target binding in silico, we selected 13 miRNA targets and their corresponding miRNA pairs to validate their interaction in vitro using miRTRAP and a luciferase reporter assay. The luciferase assays revealed that, of the 13 selected candidate SNPs, only 6 miRNA seed regions SNP in were confirmed to have all or partial loss of target binding function, while one SNP showed a novel gain of target binding in our experiment (Figure 2A). The losses of target groups were miR-940(rs65214588)/UBE2H, miR-627(rs45877714)/SMO, miR-379(rs69887451)/FLNC, miR-499(rs19786324)/CALU, miR-34a(rs23555452)/CCDC136 and miR-124(rs54878259)/TSPAN33, while the gain of target group was miR-96(rs19738442)/PLZF. These target groups were consistent with the in silico prediction. The SNP (rs19738442) in miR-96 is the first confirmed example of a novel target binding site being created by a SNP within the seed sequence. This is shown by Figure 2a — the WT miR-96 does not bind the 3’ UTR of PLZF since the relative luciferase activity is the same as the control.
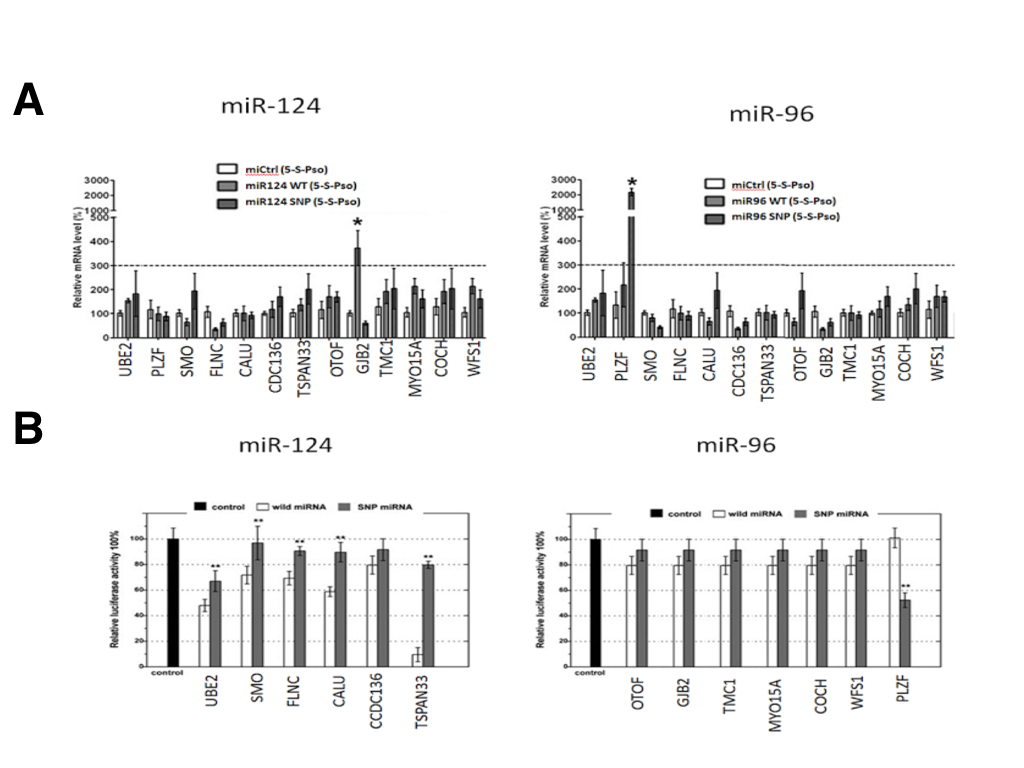
(CLICK IMAGE TO ENLARGE) Figure 2. In vitro validation of the predicted mRNA binding targets by the wild-type (white) and SNP form (grey) of miR-124 and miR-96. (A) The Luciferase reporter assay shows differential binding of one gene for both miR-124 and miR-96. The wild-type miR-24 targets the TSPAN33 transcript while the SNP-miR-24 removes this targeting. For miR-96, the wild-type does not target PLZF while the SNP causes a gain of targeting. (B) miRTRAP pull-down assays using miR-124 and miR-96. miR-124 shows moderate loss of function targeting due to SNP of the GJB2 transcripts while miR-96 shows greatly enhanced targeting ability due to the SNP of PLZF.
To further investigate whether SNPs in miRNA seed sequences could affect the interaction of miRNAs with their targets, we also performed miRTRAP pull down assay. As a surprising result, we found that only 2 of the miRNA SNPs that validated in the luciferase assay also appeared to have targeting effects in the miRTRAP analysis (Figure 2B). One miRNA SNP (miR-124; rs12349876) only modestly contributed to loss of GJB2 transcript targeting. The other SNP (miR-96 rs19738442) greatly contributed to the gain of a PLZF target. Taken together, both the luciferase and miRTRAP assay are able to validate the biological interaction of the parseltongue predisposing miR-96 variant with the PLZF transcript.
Conditional transgenesis of miR-96 in outer hair cells leads to parseltongue hyperacusis
We used a version of the tetracycline transgenesis system (TET-off) in which the tetracycline-trans-activating protein (tTA) mediates the transcription of transgenes placed under the control of the tetracycline-responsive promoter (tet-o; Figure 3A). The tTA protein itself was placed under the control of the PTRE promoter (Prestin thyroid response element), which is known to have expression restricted specifically to outer hair cells of the inner ear (Materials and Methods). Thus, the expression of all engineered transgenes was limited to OHCs and was not present in any other cell type (Figure 3B). One of two transgenes were constructed (Figure 3A). The first contained the wizard mir-96 cDNA under the control of the tetracycline-responsive promoter (tet-o-miR96). The second expressed wizard PLZF cDNA under the control of the tetracycline-responsive promoter (tet-o-PLZF). The presence of doxycycline inactivates transcription mediated by tTA (Figure 3B).
To avoid random insertion mutagenesis, targeted knock-in transgenesis was performed to target inducible expression cassettes specifically to the PLZF gene or the miR-96 gene. Targeted transgenesis was carried out using a Cre-mediated system in order to knock-in the transgenes at predetermined loci (Figure 3C). Tet-o-miR96 transgenes were knocked in at the endogenous miR-96 locus and tet-o-PLZF transgenes were knocked in at the endogenous PLZF locus. The targeted knock-in was done in order to disrupt endogenous genes so that we could ensure that all gene expression was due to our tet-off inducible system and not active endogenous genes.
We measured auditory brainstem response (ABR) hearing thresholds of “parseltongue-on” and “parseltongue-off” mice to determine when hyperacusis began in these different strains. Parseltongue-on mice had the inducible expression of the miR-96 variant (rs19738442); while, parseltongue off mice had inducible expression of the WT miR-96. Figure 4A shows the thresholds obtained from the different parseltongue predisposing conditions using tone bursts with frequencies varied from 2 to 60 kHz. For comparison, thresholds of WT non-transfected mice at 3 months age are also presented in Figure 3D (dashed line). Parseltongue-off mice showed negligible difference in hearing both low and high frequency ends, compared to the WT control, in the presence or absence of doxycycline. These mice remained unchanged between 3 months (m), 8 m and 12 m with hearing thresholds as high as 80 decibels (dB) in aged mice and as low as 60 dB in young mice (Figure 4B).This suggests that expression of the miR-96 WT allele does not correlate with parseltongue hyperacusis. Conversely, in the absence of doxycycline, parseltongue-on mice exhibited a significant reduction in their hearing threshold, even at the 60 kHz range. High-frequency hyperacusis was observed in mice as young as 3 months old and maintained throughout adulthood. Remarkably, in the absence of doxycycline, 12 m mice displayed a hearing threshold of less than 30 dB for frequencies as high as 40 kHz. In the presence of doxycycline, however, the audiogram of parseltongue-on mice resembled the WT control (Figure 4A). This shows that expression of the miR-96 SNP allele is positively correlated with the ability of mice to hear in the parselmouth frequency range.
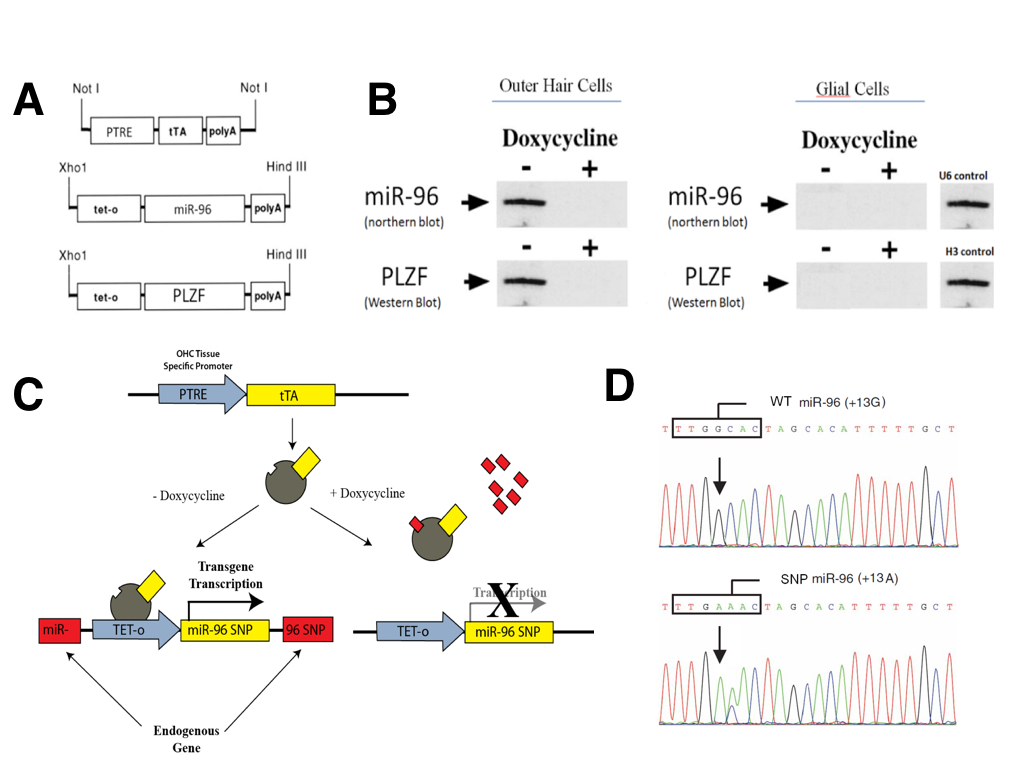
(CLICK IMAGE TO ENLARGE) Figure 3. Tetracycline dependant system to conditionally express selected transgenes in outer hair cells of the inner ear in mouse models for in vivo validation. (A) Construct of the tetracycline-trans-activating protein (tTA) under the Prestin thyroid response element (PTRE) promoter, as well as the two selected transgene-tet-o constructs (miR-96 and PLZF). (B) Northern and Western blots showing that the transgenes are only expressed in outer hair cells of the inner ear due to the PTRE promoter, and that the expression can be repressed by addition of Doxycycline. (C) Schematic representation of the TET On/Off system. (D) Sanger sequencing genotype plot depicting miR96 SNP locations.
A phenocopy experiment was done to investigate whether inducible expression of PLZF could mimic the phenotype observed in parseltongue-on mice. In the presence of the PLZF mimic mice that over-express miR-96 SNP (rs19738442) since they display increased sensitivity to frequencies in the parselmouth range (Figure 4C). By contrast, doxycycline, expression of PLZF was turned off and in the absence doxycycline nuclear accumulation of PLZF could be observed (Figure 3C). Indeed, mice with low levels of mice with active PLZF mimic mice that do not have abundant levels of miR-96 SNP (rs19738442) and concordantly do not have parseltongue hyperacusis. Taken together, it is the concomitant up-regulation of miR-96 SNP (rs19738442) and down-regulation of PLZF that seems responsible for inducing parseltongue hyperacusis in mice.
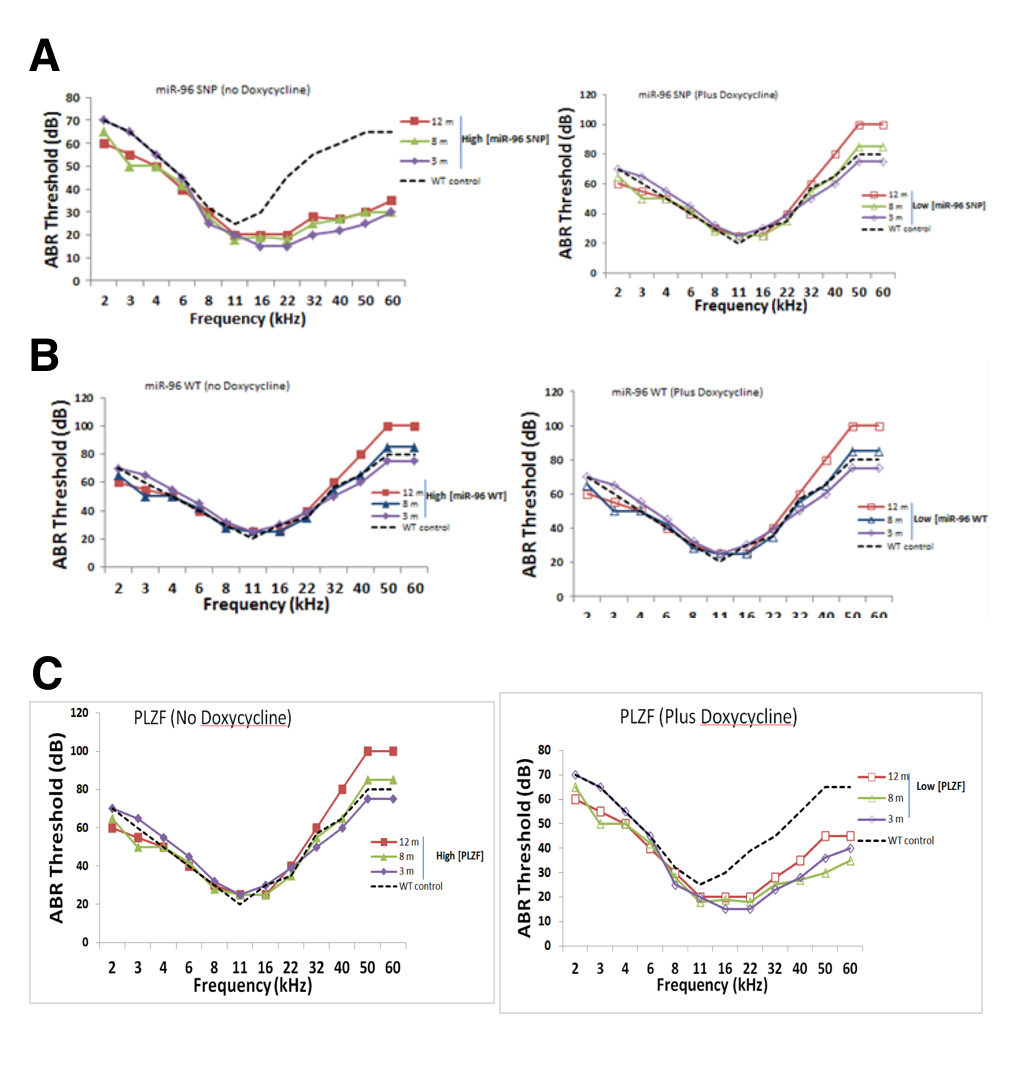
(CLICK IMAGE TO ENLARGE) Figure 4. Auditory brainstem readings (ABR) thresholds for transgenic mice at 3 monthe, 8 months and 12 months in age. A. pure tone bursts administered to parseltongue-on mice in the presence and absence of doxycycline. In the absence of doxycycline, a 30 dB threshold was maintained at 60 kHZ for mice at all stages; in the presence of doxycycline, the 30 dB threshold droped to an average of 20kHz. B. No difference in murine auditory threshold in the presence of absence of doxycycline. C. In the presence of doxycycline, mice maintain a 30dB hearing threshold of 22kHz. This 30 dB threshold is increases to an average of 40 kHz in the presence of doxycycline. Mice were administered doxycycline in their drinking water, changed once per week, at a concentration of 100 μg/ml.
Discussion
The ability to understand parselmouths has proven useful in a variety of situations and is a highly coveted attribute in most wizarding spheres [16]. Historically, studies on parseltongue speakers have been difficult to perform since many wizards choose to keep their ability hidden in an effort to avoid societal prejudice [1]. However, with the fall of the Dark Lord and increased congeniality towards parselmouths, large cohorts of parseltongue wizards with fully annotated pedigrees have only recently become available. Consequently, studies are only now being carried out to directly investigate the molecular features associated with parselmouth or parseltongue hyperacusis on a genome-wide scale. To our knowledge, a GWAS designed to assess miRNA deregulation in parseltongue hyperacusis has yet to be performed. The dataset compiled here is the largest repository of genotyping data for parseltongue and matched normal wizards generated to date (n=180 parseltongue wizards, n=3205 normal wizards).
Our Genome Wide Association Screen (GWAS) revealed 47 putative parseltongue associated or causal miRNA SNP’s. We focused our search on the miRNA seed region, as it’s the seed region that nucleates complementary mRNA target sequences and is the main determinant for miRNA targeting [17]. Generally, markers (SNPs) in a GWAS are variants used to capture association signals at particular loci and are tested as proxies for other genes that are in strong linkage disequilibrium (LD) with them. However, once genotypes are imputed, it may be interesting to predict the functional consequences of the SNPs themselves if they lie in regulatory regions such as miRNA seed sequences. To this end, two separate target prediction databases were used to identify all theoretical targets of the 47 miRNA SNPs (Materials and Methods), with positive targets being identified as interacting with at least one miRNA in both databases. The resultant mRNA target list was narrowed down by selecting targets that were differentially regulated between wild-type miRNA and SNP miRNA (Figure 1; orange and green, respectively). Gene transcripts targeted by both alleles were not considered for further analysis as these genes would likely not lead to functional consequences implicated in the parseltongue phenotype (Figure 1; blue). Targets were then filtered according to a predefined list of transcripts known to be involved in the transduction of auditory information [15]. This was done in order to enrich for miRNA-target interactions that may be involved in parseltongue hyperacuity. In total, 13 transcripts involved in the wizard auditory system were predicted to be targeted by one of two miRNAs (miR-96 and miR-124). Since these two miRNA candidates showed promising associations with the parseltongue phenotype and the auditory sensory pathway, miR-96 and miR-124 were chosen for subsequent in vitro target validation.
Luciferase reporter assays and a miRTRAP pull downs (Figure 2A and B, respectively) were used in tandem to confirm miRNA-target interactions in the biological setting and that these interactions are indeed miRNA allele specific. Consistent with in silico predictions, we found that the prestin repressor transcript, PLZF, was targeted by the miR-96 SNP (rs19738442) but not by the WT miRNA in both assays. PLZF is a transcriptional repressor which has been reported to negatively regulate the electromotility motor protein prestin in outer hair cells (OHCs) of the inner ear [18]. Numerous studies have shown that prestin is a critical protein involved in cochlear amplification and active frequency tuning in animals and wizards [5,18]. Prestin is highly expressed in OHCs with up to 10 million molecules present in the lateral membrane19. Prior work in gunea pigs has shown that OHCs responding to high frequencies have a 5-6 fold increase in prestin activity compared to low frequency OHCs [19]. Therefore, we speculate that the modulation of tonotopic prestin expression might have a physiological effect on frequency tuning within the cochlea. Given the known associations with prestin and high-frequency sound amplification, an obvious mechanism for parseltongue hyperacuity would be through its up-regulation in OHCs.
We predict that the novel interaction between miR-96 SNP (rs19738442) and the prestin repressor PLZF, alleviates transcriptional repression of prestin and leads to increased prestin levels as well as increased hearing sensitivity (Figure 5C and Figure 6B). Since serpents and parselmouths are known to speak 40 kHz range [2], it’s plausible that a surplus prestin electromtility in OHCs allows for the detection of these high frequencies; although, it still remains possible that symptoms of hyperacuity are due to some other miRNA interaction network which remains to be elucidated.
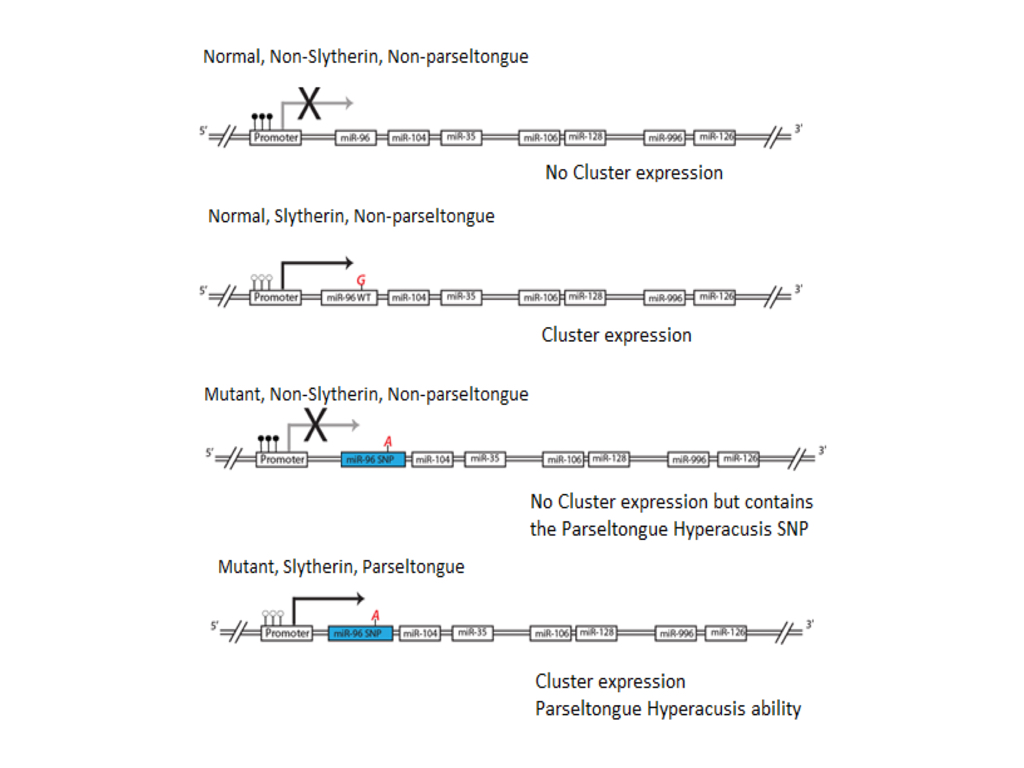
(CLICK IMAGE TO ENLARGE) Figure 5. Representation of gene cluster associated with Slytherin sorting at Hogwarts school of Withcraft and Wizardry. If the gene cluster’s promoter is methylated, whether one contains the miR-96 SNP, they will not have the parseltongue hyperacusis phenotype and will be sorted into any of the other three houses at Hogwarts (Gryffindor, Hufflepuff, and Ravenclaw). If the promoter is un-methylated then the gene cluster is expressed and one is sorted into Slytherin. If one also contains the miR-96 SNP then in addition to being sorted into Slytherin they will also have the parseltongue hyperacusis phenotype. We believe when Harry Potter was born he had the mutant miR-96 but the promoter of the gene cluster was methylated (2nd from the bottom), but that this methylation was relieved by his encounter with Lord Voldamort, changing him to have the parseltongue hyperacusis phenotype.
To test this prediction we next moved into in vivo experiments using mouse models. By using a tetracycline dependant system, we were able to generate mice that conditionally expressed a miR-96 transgene in their outer hair cells of the inner ear (Figure 3). Although auditory brainstem recording (ABR) thresholds of mice have been previously examined in muggle studies [20-22], we used a wider range of frequencies to determine thresholds at higher frequencies characteristic of the parselmouth language. As predicted, expression of the mir-96 SNP transgene led to cytomplasmic accumulation of Prestin and parseltongue hyperacusis. The ABR audiogram indicated that miR-96 SNP mice in the absence of doxycycline (parseltongue-on mice) display early onset and rapid progression of hyperacusis as early as 3 months of age. Inactivation of the miR-96 SNP transgene prevented development of parseltongue hyperacusis and caused sustained reduction of cytoplasmic Prestin (Figure 4A). These parseltongue-off mice (miR-96 SNP mice in the presence of doxycycline) display classical high-frequency prebycusis. Regression of parseltongue hyperacusis apparently resulted from a decrease in miR-96 levels and concordantly increased PLZF activity. Next we wanted to confirm whether the parseltongue hyperacusis phenotype observed in parseltongue-on mice was mediated by targeting of the prestin repressor. Remarkably, mice in the presence of doxycycline displayed decreased expression of PLZF (Figure 3B) and were able to phenocopy parseltongue hyperacusis observed in parseltongue-on mice (Figure 4C). These data indicate that the specific interaction of mir-96 SNP (rs19738442) with PLZF, the prestin repressor transcript, is sufficient for the induction of parseltongue hyperacusis.
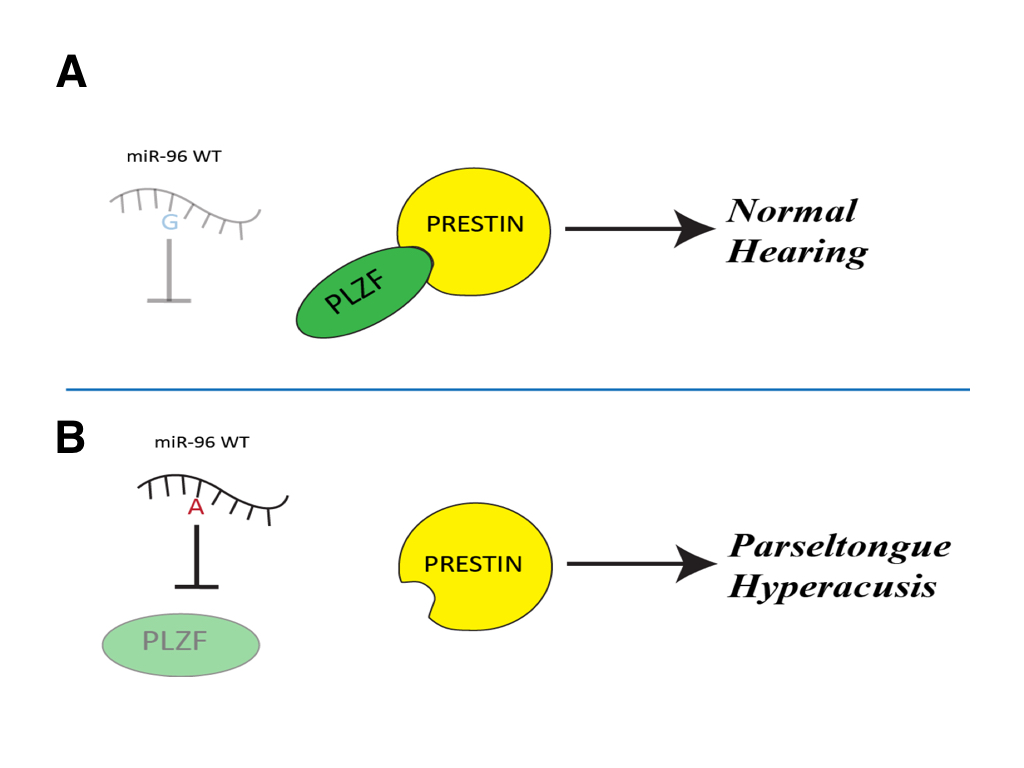
(CLICK IMAGE TO ENLARGE) Figure 6. A schematic representation of predicted path for miR-96 with and without the G-A SNP. (A) The wild-type miR-96 is unable to recognize the PLZF mRNA, so PLZF is able to be transcribed and bind to Prestin inhibiting its activity. This results in regular sized outer hair cells of the inner ear and you get regular hearing. (B) If miR-96 with the G-A SNP is able to be expressed it is able to bind and repress PLZF mRNA so that it is not transcribed. This causes Prestin to be activated and causes longer outer hair cells of the inner ear, resulting in the ability to hear frequencies in the 40-50kHz range and therefore given one the parseltongue hyperacusis phenotype.
Lastly, we observed that miR-96 mapped to a single polycistronic transcript on chromosome 7q32 that was reported to be expressed in the inner ear [23]. Since parseltongue individuals are almost unanimously sorted into the Slytherin house at Hogwarts School of Witchcraft and Wizardry, we wondered whether expression of this 7q32 miRNA gene cluster was correlated with sorting into any of the houses at Hogwarts. To this end, a total of 438 samples were downloaded from The Wizard Genome Atlas Project [24] (TWGA) and sorted according to their house annotation. Interestingly, we found that high-expression of the miR-96 gene cluster was positively correlated with Slytherin sorting status (Figure 7B; anova, p<0.0001). Since the Slytherin subtype is enriched for expression of miR-96 gene cluster, we speculate that this polycistronic transcript plays a key role in the regulation of house placement outcome by the sorting hat. Moreover, we observed that methylation of this gene cluster’s transcriptional start site (TSS; Cg07388018) was negatively correlated with expression (Figure 7A). Taken together, these data indicate that DNA methylation is a mechanism by which the 7q32 miRNA cluster might be turned on or off and that the resultant expression status of this cluster is important for Slytherin classification.
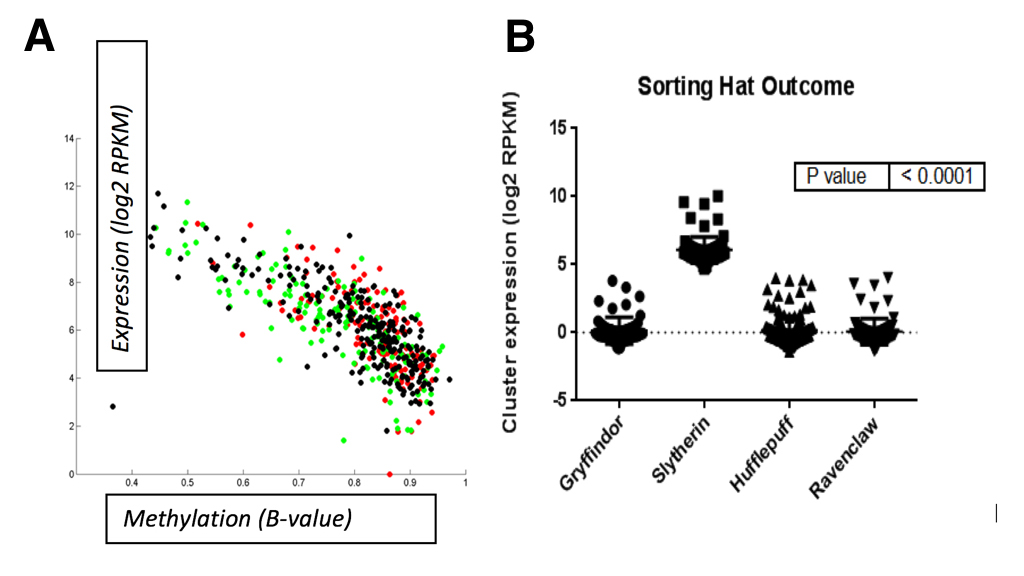
(CLICK IMAGE TO ENLARGE) Figure 7. A. Methylation status for a TSS probe (Cg07388018) of the polycistronic miRNA gene cluster is negatively correlated with miRNA expression. Spearman correlation was performed in matlab; R=0.89, p=0.006. Correlation B. High-expression of the miR-96 gene cluster was positively correlated with Slytherin sorting status (anova; p<0.0001). Correlation was performed using GraphPad software.
Interestingly, the fact that this polycistronic gene cluster might be epigenetically regulated provides an elegant model for explaining how Harry Potter could have been endowed with the ability to speak parseltongue after Lord Voldemort used the killing curse on him (Figure 6). It is probable that Harry was a carrier of the miR-96 G-A SNP (rs19738442) at birth but the polycistronic gene cluster encompassing this SNP was hypermethylated at the promoter region and not expressed. Upon performing the unforgivable curse (Avada Kedavara), Lord Voldemort unknowingly relieved methylation of the hitherto silenced 7q32 gene cluster and allowed the expression of the parseltongue hyperasusis miR-96 SNP. Expression of this gene cluster may also explain why the sorting hat said that Harry would have done well in Slytherin. Given the preliminary implications of this gene cluster in Slytherin sorting, external validation and further investigation into the molecular function of this gene cluster is warranted.
Conclusion
In summary, our study describes a method to relate miRNA SNPs to parseltongue-associated SNPs from GWAS, for the purpose of identifying parseltongue hyperacusis-suceptibility miRNA SNPs. Through computational methods and in vitro target validation, we were able to discover a unique SNP variant in miR-96, which targeted and repressed of the PLZF prestin repressor transcript. The prestin repressor is involved in regulating the expression of a network of genes involved in OHC electromotility. Thus, the parseltongue miR-96 SNP may result in increased parseltongue hyperacusis by affecting miRNA-based regulation of the prestin repressor. We conclude that, despite the multistep nature of parseltongue hyperacusis, remediation of a single genetic lesion can induce sensitivity to serpent frequencies and might have important therapeutic value.
Conflict of interest
Authors have no conflicts of interest to declare
Acknowledgements
Funding support for this work is through research grants to J.L.C and D.A.R from Institutes for Wizard Research (IWR), British Wizard Society, and the Ministry of Magic.
Author’s contribution
J.L.C and D.A.R participated in study concept and design; acquisition of data; analysis (including statistical analysis) and interpretation of data; and drafting of the manuscript. D.A.R participated in data acquisition, preprocessing and analysis. J.L.C participated in drafting of the manuscript and its critical revision.
References
1. Parseltongue. (n.d.) Retrieved from: http://harrypotter.wikia.com/wiki/Parseltongue
2. Ekans S. and Arboc S. (2013). Identification of a high frequency noise from snakes. Erutan, 7(6), 15-26
3. Anandaram, H., & Anand, D. A. A Computational approach for identifying novel micro RNAs from Genome Wide Association Studies of Psoriasis.
4. Sethupathy, P., & Collins, F. S. (2008). MicroRNA target site polymorphisms and human disease. Trends in genetics, 24(10), 489-497.
5. Botts, B. (2012). Two unique miRNA’s (24 and 48) differently regulated in Squibs. Wizardry, 52(13), 555-556.
6. Richardson, K., Lai, C. Q., Parnell, L. D., Lee, Y. C., & Ordovas, J. M. (2011). A genome-wide survey for SNPs altering microRNA seed sites identifies functional candidates in GWAS. BMC genomics, 12(1), 504.
7. Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 116(2), 281-297.
8. Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell, 136(2), 215-233.
9. Iwai, N., & Naraba, H. (2005). Polymorphisms in human pre-miRNAs. Biochemical and biophysical research communications, 331(4), 1439-1444.
10. Ryan, B. M., Robles, A. I., & Harris, C. C. (2010). Genetic variation in microRNA networks: the implications for cancer research. Nature Reviews Cancer, 10(6), 389-402.
11. Landi, D., Gemignani, F., Barale, R., & Landi, S. (2008). A catalog of polymorphisms falling in microRNA-binding regions of cancer genes. DNA and cell biology, 27(1), 35-43.
12. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. (2011). Genome-wide association study identifies five new schizophrenia loci. Nature genetics, 43(10), 969-976.
13. Gong, J., Tong, Y., Zhang, H. M., Wang, K., Hu, T., Shan, G., … & Guo, A. Y. (2012). Genome‐wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Human mutation, 33(1), 254-263.
14. Marchini, J., & Howie, B. (2010). Genotype imputation for genome-wide association studies. Nature Reviews Genetics, 11(7), 499-511.
15. Alsaber R, Tabone CJ, Kandpal RP. (2006) Predicting candidate genes for human deafness disorders: a bioinformatics approach. BMC Genomics, 19;7:180.
16. Myrtle, M., Nick, N. H., Friar, F., Lady, G., Baron, B., & Binns, C. (2010). 1000 years of Hogwarts History from within the Walls. Protoplasm, 25(100), 1192-1194.
17. Grimson, A., Farh, K. K. H., Johnston, W. K., Garrett-Engele, P., Lim, L. P., & Bartel, D. P. (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Molecular cell, 27(1), 91-105.
18. Hudspeth AJ. (2014). Integrating the active process of hair cells with cochlear function. Nat Rev Neurosci. Sep;15(9):600-14.
19. Bai, J. P., Surguchev, A., Ogando, Y., Song, L., Bian, S., Santos-Sacchi, J., & Navaratnam, D. (2010). Prestin surface expression and activity are augmented by interaction with MAP1S, a microtubule-associated protein. Journal of Biological Chemistry, 285(27), 20834-20843.
20. Spongr, V. P., Flood, D. G., Frisina, R. D., & Salvi, R. J. (1997). Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. The Journal of the Acoustical Society of America, 101(6), 3546-3553
21. Willott, J. F., Parham, K., & Hunter, K. P. (1991). Comparison of the auditory sensitivity of neurons in the cochlear nucleus and inferior colliculus of young and aging C57BL/6J and CBA/J mice. Hearing research, 53(1), 78-94.
22. Sha, S. H., Kanicki, A., Dootz, G., Talaska, A. E., Halsey, K., Dolan, D., … & Schacht, J. (2008). Age-related auditory pathology in the CBA/J mouse. Hearing research, 243(1), 87-94.
23. Mencía, Á., Modamio-Høybjør, S., Redshaw, N., Morín, M., Mayo-Merino, F., Olavarrieta, L., … & Moreno-Pelayo, M. Á. (2009). Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nature genetics, 41(5), 609-613.
24. The Wizard Atlas Genome Project
25. Lin, S., Carvalho, B., Cutler, D., Arking, D. E., Chakravarti, A., & Irizarry, R. A. (2008). Validation and extension of an empirical Bayes method for SNP calling on Affymetrix microarrays. Genome Biol, 9(4), R63.
26. Baigude, H., Li, Z., Zhou, Y., & Rana, T. M. (2012). miR‐TRAP: A Benchtop Chemical Biology Strategy to Identify microRNA Targets. Angewandte Chemie, 124(24), 5982-5985.
27. Choi, Y. J., Son, M. Y., & Hasty, P. (2011). One‐step knockin for inducible expression in mouse embryonic stem cells. Genesis, 49(2), 92-97.
28. Li, M., Tian, Y., Fritzsch, B., Gao, J., Wu, X., & Zuo, J. (2004). Inner hair cell Cre‐expressing transgenic mouse. genesis, 39(3), 173-177.
29. Kim, T. M., Choi, Y. J., Ko, J. H., & Hasty, P. (2008). High‐throughput knock‐in coupling gene targeting with the HPRT minigene and Cre‐mediated recombination. genesis, 46(12), 732-73.
30. Song, L., McGee, J., & Walsh, E. J. (2008). Development of cochlear amplification, frequency tuning, and two-tone suppression in the mouse. Journal of neurophysiology, 99(1), 344-355.